


A common genetic mutation that helps some kidney cancers survive may also expose an unexpected weakness, one that MUSC Hollings Cancer Center researchers hope to transform into a new treatment strategy.
In a new study published in Cancer Research, Aguirre de Cubas, Ph.D., and colleagues discovered that kidney cancer cells lacking the tumor suppressor gene SETD2 become highly dependent on a protein called BCL-xL for survival. By targeting that dependency, the researchers were able to selectively eliminate SETD2-deficient cancer cells in laboratory models while largely sparing cancer cells with intact SETD2. The findings identify a potential new therapeutic strategy for patients with a particularly aggressive subset of kidney cancers.
That discovery laid the foundation for a newly awarded five-year, multi-million-dollar National Cancer Institute (NCI) grant. The grant will support efforts to understand why SETD2-deficient tumors become dependent on BCL-xL and whether that vulnerability can be leveraged to develop more effective treatments.
"These projects are two parts of the same scientific journey," de Cubas said of the study and grant. "The study revealed a previously unrecognized vulnerability in SETD2-mutant kidney cancers. The grant now gives us the opportunity to understand the biology behind that vulnerability and whether it can be translated into a new therapeutic strategy for patients."
A common mutation with unanswered questions
SETD2 is one of the most frequently altered tumor suppressor genes in human cancer, with alterations occurring in approximately 5% of all solid malignancies. It is particularly common in clear cell renal cell carcinoma, which accounts for almost three-quarters of all kidney cancers and is difficult to treat once it spreads beyond the kidney.
Researchers estimate that roughly 20% to 25% of tumors lose the gene entirely, and patients whose tumors carry SETD2 alterations often experience more aggressive disease and poorer outcomes. Despite its prevalence and clinical importance, researchers have struggled to understand why losing SETD2 confers a selective advantage to kidney tumors.
What we uncovered was essentially an Achilles’ heel. By losing SETD2, these tumors gain certain advantages that help them grow, but they also become highly dependent on BCL-XL for survival. That dependency creates a therapeutic opportunity.
"The central question was why tumors experience loss of SETD2 in the first place,” de Cubas said. “SETD2 is one of the most frequently mutated genes in kidney cancer, yet we don’t fully understand what advantages its loss provides to tumor cells. Understanding that biology could reveal vulnerabilities that can be exploited therapeutically, which is important for patients with limited treatment options."
To answer that question, his team searched for what cancer biologists call "synthetic lethal vulnerabilities,” or hidden weaknesses that emerge when a cell acquires a cancer-promoting mutation. In these cases, the mutation creates an unexpected dependence on another gene or protein for survival. By targeting that dependency, researchers can selectively eliminate cancer cells while leaving normal tissues largely unharmed, creating opportunities to develop more precise and effective therapies or improve existing ones.
Their search revealed a dependency on BCL-xL, a protein that acts as a cellular bodyguard by preventing programmed cell death. While most cells can survive without relying heavily on BCL-XL, kidney cancer cells lacking SETD2 had become remarkably dependent on it.
When the researchers blocked BCL-xL, the results were striking. SETD2-deficient cancer cells died rapidly, whereas kidney cancer cells with intact SETD2 remained largely unaffected.
"It was an unexpected finding," de Cubas said. "What we uncovered was essentially an Achilles’ heel. By losing SETD2, these tumors gain certain advantages that help them grow, but they also become highly dependent on BCL-xL for survival. That dependency creates a therapeutic opportunity."
DNA where it shouldn't be
Investigating that vulnerability led researchers to the mitochondria.
Often described as the cells’ powerhouses, mitochondria contain their own DNA separate from the genetic material housed in the cell nucleus. Under normal circumstances, that DNA remains safely confined within the mitochondria. But the researchers found that cells lacking SETD2 experienced mitochondrial stress that caused small amounts of DNA to leak into the cell's interior.
That leakage raised cellular alarm bells. Because, according to de Cubas, "DNA is not supposed to be floating around in the cytoplasm. When the cells detect DNA where it shouldn't be, they interpret it as a danger signal, often resembling a viral infection.”
The leaked mitochondrial DNA activated a molecular pathway known as cGAS-STING, which serves as part of the body’s innate immune defense system that evolved to detect viral pathogens. The result was a persistent inflammatory state that fundamentally changed the biology of SETD2-deficient cancer cells.
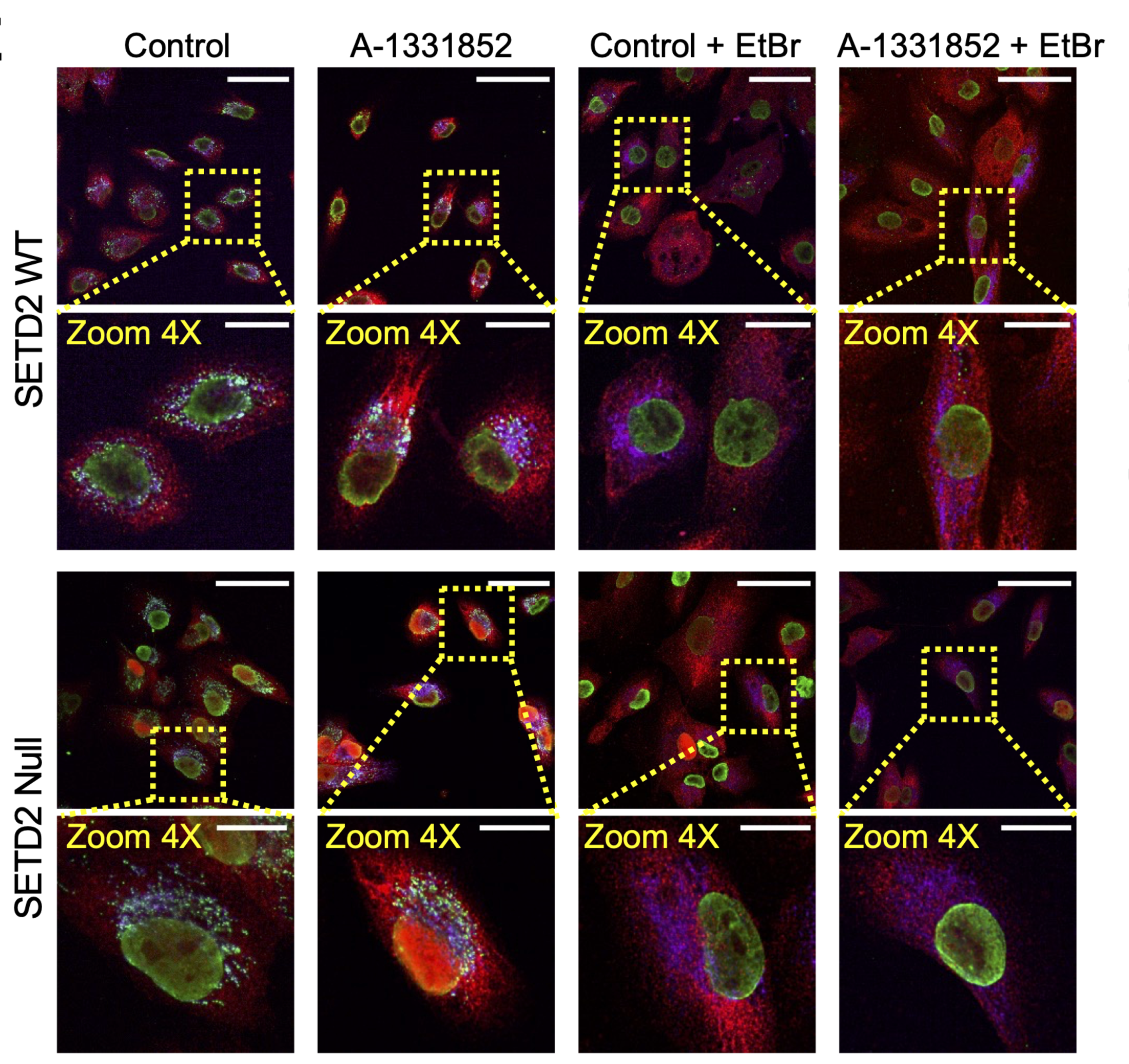
This figure uses immunofluorescence microscopy to show where different components are located inside the cells after different treatments. The goal is to determine whether A-1331852 (BCL-xL inhibitor) causes mitochondrial DNA release that activates the inflammatory transcription factor IRF3.
The first two rows show wild-type SETD and the second two rows are SETD2-deficient.
The columns are the treatment conditions:
Control = no drug (baseline).
A-1331852 = BCL-xL inhibitor (tests whether blocking BCL-xL activates inflammatory signaling).
Control + EtBr = ethidium bromide only (ethidium bromide depletes mitochondrial DNA, testing whether mtDNA is required).
A-1331852 + EtBr = BCL-xL inhibitor + mtDNA depletion (tests whether the effects of A-1331852 disappear when mtDNA is removed).
"When we treated the cells with BCL-xL inhibitors, we amplified an inflammatory program that was already smoldering," de Cubas said. "The cells essentially behaved as though they were responding to a viral infection, pushing them toward a point they could no longer tolerate."
The effect was twofold. BCL-xL inhibition drove cancer cells toward self-destruction while simultaneously increasing inflammatory signals that made them more visible to the immune system.
"What makes this particularly exciting is that the biology appears to be interconnected," de Cubas said. "The same mitochondrial stress that helps create the vulnerability may also contribute to immune activation, potentially giving us multiple ways to exploit the weakness therapeutically."
Solving a kidney cancer mystery
The findings also help to solve one of kidney cancer's longstanding mysteries.
Unlike cancers such as melanoma or lung cancer, kidney tumors often respond surprisingly well to immunotherapy despite carrying relatively few mutations that would be expected to make them visible to the immune system. During the study, de Cubas and colleagues found evidence that mitochondrial DNA leakage may contribute to the inflammatory signal that makes kidney tumors immunologically active.
"We think mitochondrial DNA may have a much more central role in kidney cancer immunogenicity than previously appreciated,” de Cubas said.
The discovery also challenges prevailing assumptions about the source of inflammatory signaling in SETD2-deficient tumors. Because the loss of SETD2 is known to cause chromosomal instability and the formation of micronuclei – small fragments of misplaced nuclear DNA that can activate immune pathways – many researchers had assumed that micronuclei were the primary trigger of inflammation in these cancers.
Instead, de Cubas' team found that mitochondrial DNA leakage was a much stronger driver of cGAS-STING activation.
We think mitochondrial DNA may have a much more central role in kidney cancer immunogenicity than previously appreciated.
"The field has largely focused on micronuclei as the source of inflammatory signaling in SETD2-deficient cells," de Cubas said. "What we found is that mitochondrial DNA leakage appears to be the dominant signal. The micronuclei were there, but they may have been masking a much more important mechanism."
The findings may also help to resolve broader questions about what drives immune activation in kidney tumors. The new work points to mitochondrial DNA leakage as a central initiator of inflammatory pathway responses.
From discovery to therapy
The newly awarded NCI grant will build directly on these findings by pursuing two fundamental questions. de Cubas and his team will investigate what causes mitochondrial DNA leakage in SETD2-deficient tumors and whether that process can be exploited to make tumors more susceptible to treatment.
The researchers will also investigate whether combining BCL-xL inhibition with immune checkpoint blockade therapies can produce a more potent anti-tumor response. Because BCL-xL inhibition appears to amplify antiviral-like inflammatory signaling in SETD2-deficient tumors, the combination could make these cancers more responsive to immunotherapy.
The work also highlights the collaborative nature of research at Hollings. de Cubas credited co-authors Tim Barnoud, Ph.D., Jezabel Rodriguez Blanco, Ph.D., and Thai Ho, M.D., Ph.D., with helping to shape the project and refine its scientific direction. The study also relied heavily on the services and capabilities of Hollings’ Flow Cytometry and Cell Sorting and Cell and Molecular Imaging shared resources.
Although the findings remain preclinical, de Cubas believes they provide a strong foundation for future translational studies. The new award will allow his laboratory to expand its research program and recruit additional post-doctoral fellows and research staff to pursue these questions.
"If we can overcome some of the challenges associated with targeting BCL-xL, the effect we see in SETD2-mutant tumors is remarkably strong," he said. "We now have a clear path forward for understanding the underlying tumor biology and, most importantly, whether we can use that knowledge to develop better therapies for patients."
Featured in this story

Aguirre De Cubas, Ph.D.
Dr. De Cubas' research program is focused on 1) understanding how cancers interact with the immune system, 2) identifying molecular determinants of tumoral immunogenicity and antigenicity, and 3) understanding how commonly lost epigenetic modifiers (SETD2, PBRM1, and BAP1) modify the tumor microenvironment and contribute to progression of clear cell renal cell carcinoma (ccRCC).
To accomplish these goals, the research program operates at both dry and wet lab levels and brings a balanced skillset in genomics and bioinformatics, as well as cancer biology and immunology. Successful completion of this research will contribute to our knowledge of the mechanisms behind response to ICB in ccRCC. These novel discoveries would provide critical biological insights that ultimately could lead to novel clinical treatment strategies for ccRCC.

Thibaut 'Tim' Barnoud, Ph.D.
Education and Training
2016-2021 Postdoctoral Fellow, Molecular and Cellular Oncogenesis Program, The Wistar Institute
2010-2015 Ph.D., Department of Biochemistry and Molecular Genetics, University of Louisville
2005-2009 B.A., Department of Biology, University of Louisville
Research
Dr. Barnoud’s laboratory uses genetically engineered mouse models (GEMMs) to study the p53 tumor suppressor, the most frequently mutated gene in human cancer. Their primary focus is on the impact of p53 variants on cancer risk and response to therapy, primarily centered on colorectal cancer. Mechanistically, their research aims to determine the influence of the African-centric P47S variant of p53 on colorectal cancer progression using genetically engineered cell lines, transgenic and colitis-induced mouse models of cancer. His lab is also actively investigating the role of another African-centric variant of p53, Y107H. The long-term goal of their research program is to leverage genetic variants of p53 in order to better understand the role of p53-mediated tumor suppression.
A second major research focus of his laboratory is to understand the role of heat shock proteins (HSPs) on the initiation and progression of cancer. They have studied specific inhibitors that target HSPs with the hopes of developing effective therapies for gastrointestinal (GI) cancers. Specifically, his laboratory is focused on novel small-molecule inhibitors of HSP70 as a potential therapeutic target for GI cancers. They found that a significant fraction of HSP70 localizes to the mitochondria of tumor cells; as a result, they generated novel mitochondrial-targeting HSP70 inhibitors. They are leveraging these inhibitors in order to better understand the role of HSP70 in the initiation, progression, and metastasis of GI cancers. They are also interested in identifying novel combination strategies of HSP70i with standard of care (i.e., Gemcitabine, immune checkpoint inhibitors) in order to improve.

Thai Ho, M.D., Ph.D.
Dr. Thai Ho is an oncologist specializing in treating genitourinary cancers (bladder, upper tract, kidney). In the lab, Dr. Ho investigates clear cell renal cancer (ccRCC), which is among the top 10 leading causes of cancer deaths in the United States. He uses SETD2 mutations in ccRCC as a paradigm for investigating the mechanisms that underpin how these mutations drive cancer progression and to look for novel ways to target these mutations, which are still mostly regarded as undruggable.

Jezabel Rodriguez-Blanco, Ph.D.
My research is focused on the most common malignant pediatric brain tumor, medulloblastoma, and aimed at developing novel therapeutics for underserved patients suffering from this cancer. Because of the significant morbidity that current multimodal therapies have on the developing brains of children, one of my major research lines is focused on finding less toxic targeted therapeutics. Unfortunately, tumors exposed to single agent targeted therapeutics tend to recur. As virtually none of the children whose tumors relapse will live beyond one year, it is critical to identify the mechanisms allowing medulloblastoma to grow back, and design therapeutics aimed at targeting them.
Reference
Anusha Uprety, Chandler Judd, Emily D. Villella, Rebecca Keller, Ryan Wagner, Keith D. Robertson, Jezabel Rodriguez-Blanco, Thibaut Barnoud, Frank M. Mason, Thai H. Ho and Aguirre A. de Cubas. SETD2 Deficiency Drives Mitochondrial DNA Leakage and Creates a Druggable Dependency on BCL-xL in Clear Cell Renal Cell Carcinoma. Cancer Research [2 June 2026]. doi: 10.1158/0008-5472.CAN-25-3195.
Grants from the Department of Defense (HT9425-24-1-0613), National Cancer Institute (K01CA245431; R01CA271503; R01CA275082), National Institute of Neurological Disorders and Stroke (R01NS138021; K01NS119351) and National Institute of General Medical Sciences (P20GM130457) supported this research. Funding also came from an MUSC COBRE in Digestive and Liver Disease sub-award and an Alex’s Lemonade Stand Foundation for Childhood Cancer “A” Award (23-28298).

Hayley Kamin
Communications Manager
Hayley Kamin is the communications manager for the Hollings Cancer Center Communications and Marketing team, having joined the team in 2025 after three years as a communications specialist at the National Institutes of Health (NIH). As a science communicator with a Ph.D. from the University of Florida, she has extensive experience translating complex research into clear, engaging content. Her career has included roles at the NIH’s National Institute of Mental Health and the American Psychological Association, where she led content development and editorial strategy, developed science and health communications and worked with researchers and clinicians to strengthen public understanding of research.
Contact Hayley at kamin@musc.edu.
Recent Cancer Research stories


